中国病毒学论坛|我们一直在坚持!
标题: Scientific reports:H7N9禽流感病毒感染与肠道菌群的关系 [打印本页]
作者: ipsvirus 时间: 2015-11-10 13:05
标题: Scientific reports:H7N9禽流感病毒感染与肠道菌群的关系
本帖最后由 ipsvirus 于 2015-11-10 13:07 编辑

2013年首次在中国出现人感染H7N9病毒病例,一时间引发了全国各地人们对禽流感的恐慌。
H7N9为什么如此可怕?H7N9亚型禽流感病毒是甲型流感中的一种,以前仅在禽间发现,后来出现在人类身上,临床上人感染H7N9潜伏期为7天以内,患者一般表现为流感样症状,如发热,咳嗽,少痰,可伴有头痛、肌肉酸痛和全身不适。重症患者病情发展迅速,表现为重症肺炎,体温大多持续在39摄氏度以上,出现呼吸困难,可伴有咳血痰;可快速进展出现急性呼吸窘迫综合征、纵隔气肿、脓毒症、休克、意识障碍及急性肾损伤等。
近日,浙江大学传染病国家重点实验室及感染性疾病协同创新中心的李兰娟院士、秦楠研究员及郑焙文、姚坚博士等团队人员采用宏基因组技术对H7N9患者肠道菌群进行了研究,文章发表于Nature集团子刊《Scientific Reports》。研究分析了抗生素治疗对肠道菌群产生的影响。该项研究结果寻找的与H7N9患者密切相关的特异性微生物标记物为指导临床用药以及研发益生菌和检测试剂盒提供了理论依据。
研究者们选取了26例患者的93个粪便样品为研究对象,第一次取样时间为入院当天, 26个患者中,17个接受了抗生素治疗(AB),9个受非抗生素治疗(NAB),平均年龄57岁。利用Illumina Hiseq2500 PE100bp测序平台进行宏基因组测序,平均每个样品产生2.9Gb数据。然后选取31个健康人肠道样品测序数据与26例患者病人肠道样品测序数据进行了比较分析,并与HMP计划,MetaHit计划及T2D三个基因集进行了比较分析。分析结果发现H7N9基因集中有528, 592个独特基因。
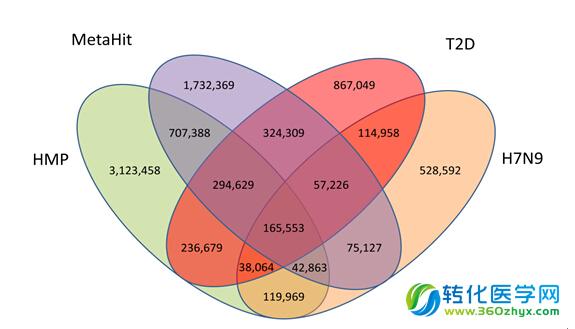
图1 韦恩图显示当前主要人类肠道微生物基因集的关系
为了研究抗生素对患者肠道菌群的影响,比较了AB和NAB肠道菌群中的Top 20物种,发现大肠杆菌(Escherichia coli)在AB和NAB患者中丰度分别为第一和第三,说明Escherichia coli作为一种潜在的致病菌,其丰度与H7N9的感染和治疗过程有关。除Escherichia coli外,粪肠球菌Enterococcus faecium和肺炎克雷伯氏菌Klebsiella pneumoniae也在病人中富集。柔嫩梭菌Faecalibacterium prausnitzii在AB中显著降低。
为研究肠道微生物在患者肠道与健康人肠道中的区别,将健康人和患者肠道微生物进行了比较。通过MGS(宏基因组物种)分析,健康人中富集的是罗氏菌(Roseburia inulinivorans DSM 16841)、产丁酸盐细菌(bacteriumSS3/4)、真杆菌(Eubacterium ventriosum ATCC 27560)、罗氏弧菌(Roseburia intestinalis)和瘤胃球菌(Ruminococcus);H7N9患者中富集的是梭菌(Clostridium sp. 7 2 43FAA)、场球菌(Enterococcus)、肠杆菌(Enterobacter)和丁酸梭菌(Clostridium butyricum)。
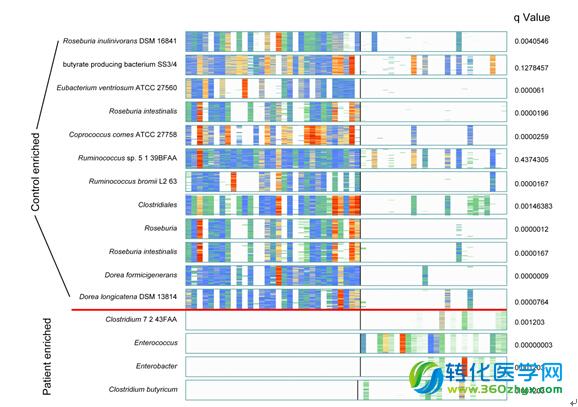
图2 健康人和患者MGS差异比较
另外,由多样性指数看出,患者和健康人相比,肠道微生物物种多样性下降,而受抗生素的治疗的患者下降的更为严重。肠型分析结果表明两种肠型主要驱动菌属为拟杆菌(Bacteroides)和普氏菌(Prevotella),第三种肠型的属为克雷伯氏菌(Klebsiella)且只包含H7N9病人,说明H7N9病毒的入侵导致病人肠型的改变,而健康人中肠道菌群结构稳定。
KEGG分析结果表明四个基因集共有基因丰度最高的主要是酶家族,其次是膜转运和碳水化合物代谢;eggNOG分析结果表明四个基因集共有基因丰度最高的主要是一些功能未知的基因,其次是氨基酸转运和代谢相关基因。
综上所述,宏基因组分析结果告诉我们:抗病毒物质,益生菌及抗生素治疗可以提高H7N9患者肠道有益菌的微生物多样性及丰度。
来源:转化医学网
作者: ipsvirus 时间: 2015-11-10 13:09
Influence of H7N9 virus infection and associated treatment on human gut microbiota
Nan Qin, Beiwen Zheng, Jian Yao, Lihua Guo, Jian Zuo, Lingjiao Wu, Jiawei Zhou, Lin Liu, Jing Guo, Shujun Ni, Ang Li, Yixin Zhu, Weifeng Liang, Yonghong Xiao, S. Dusko Ehrlich & Lanjuan Li
Between March and June, 2013, forty H7N9 patients were hospitalized in our hospital. Next-generation sequencing technologies have been used to sequence the fecal DNA samples of the patient, the within sample diversity analysis, enterotyping, functional gene and metagenomic species analysis have been carried on both the patients and healthy controls. The influence of associated treatment in H7N9 infected patients is dramatic and was firstly revealed in species level due to deep sequencing technology. We found that most of the MetaGenomic Species (MGS) enriched in the control samples were Roseburia inulinivorans DSM 16841, butyrate producing bacterium SS3/4 and most of MGS enriched in the H7N9 patients were Clostridium sp. 7 2 43FAA and Enterococcus faecium. It was concluded that H7N9 viral infection and antibiotic administration have a significant effect on the microbiota community with decreased diversity and overgrowth of the bacteria such as Escherichia coli and Enterococcus faecium. Enterotype analysis showed that the communities were unstable. Treatment including antivirals, probiotics and antibiotics helps to improve the microbiota diversity and the abundance of beneficial bacteria in the gut.
http://www.nature.com/articles/srep14771
| 欢迎光临 中国病毒学论坛|我们一直在坚持! (http://virology.com.cn/) |
Powered by Discuz! X3.2 |